咨询热线
13701143136
13701143136

追求合作共赢
Win win for you and me售前售中售后完整的服务体系
诚信经营 质量保障 价格合理 服务完善当前位置:首页 > 雷竞技raybet网站即时 > 薄层色谱成像分析仪中点样技术应用方案-如何点样
薄层色谱成像分析仪中不同溶剂对样品的洗脱能力不同,因此在点样的同时,样品在原点开始呈环形展开,如果样品在溶剂中的溶解度很大,原点将变成空心环,Kaiser称这种现象为“点样环形色谱效应”。这种效应对随后的线形展开造成不良影响,因此配制样品溶液时应选用对组分溶解度相对较小的溶剂;溶剂的粘度不宜过高,以便于点样,溶剂的沸点要适中,沸点过低由于挥发会改变样品溶液浓度导致较大误差,沸点过高,样品溶液的溶剂会在原点残留,导致改变展开剂的选择性,特别是样品溶液的溶剂与展开剂的极性相差较大时更为明显,zui常用的溶剂为甲醇及乙醇,有时也用丙酮。点样后必须将溶剂全部除去后再进行展开,但要避免高温加热,以免改变待测成分的性质。
点样体积经典薄层一般为1-5μl,薄层为100-500nl,样品溶液浓度一般在0.01%-1.00%范围内,点样过多会造成原点“超载”,展开剂产生“绕行”现象使斑点拖尾或重叠,使扫描峰形不对称或不能基线分离,并由于超载得到非线性的校正曲线,严重影响定量结果,降低准确度。
经典薄层的原点直径zui大不得超过5mm,一般3mm较为合适,薄层原点直径约为1-2mm,尽可能避免多次点样,原点直径过大会降低分辨率及分离度。
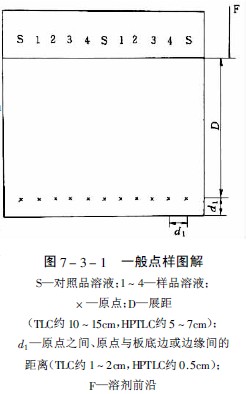
经典薄层色谱成像分析仪中薄层起始线距底边约1.5cm,薄层为1cm,展距前者为10-15cm,后者为5-7cm,对于难分离的化合物用多元展开剂展开时,注意原点与展开剂的距离每次要保持一致,否则由于色谱分离zui初行为的差异而导致分离失败。
点与点的间距经典薄层为1-2cm,薄层为0.5cm,用具自动换行功能的薄层扫描仪时,斑点之间的距离必须十分,才能正确地扫描每行的色斑,得到好的测定结果,图7-3-1为一般的点样图解。
在点样前,应将薄层板在日光及紫外光下检查板面有无损坏或污染,选择合格的薄层板后决定用点状点样还是用带状点样。为此,瑞士CAMAG公司生产了系列点样设备以供选用,这些设备只适用于硬板。下面是常用的点样方式及设备。 北京威锋技术开发公司
(一)点状点样
如果薄层色谱成像分析仪仅仅为了定性,用内径约0.5mm管口平整的毛细管或微量注射器将样品溶液点在距薄层底边约2cm处,点样直径不超过5mm,点间距约为1-1.5cm即可。
为进行定量,借毛细作用吸样的定容管有两种,一是容积为0.5、1、2、3、4及5μl的定量毛细管(Microcaps),另一种是100及200nl的铂铱合金定量毛细管(Nanopipette);注射器式的可变体积的点样器有50-230nl的毫微点样器(Nano-Applicator)及0.5-2.3μl的微量点样器(Micro-Applicator),用于需要调节体积及没有毛细作用的键合相薄层的点样。
毛细管式的微量点样器装在手动的Nanomat型点样装置的磁性点样头上,点样头装在弹簧上。按下此点样头,毛细管借恒定的压力接触薄层表面。点间距离由带准确的#44 间隔的E字形缺刻装置控制,适用于普通或薄层板,点样体积,样品原点小,原点间隔定位准确;注射器式的毫微点样器及微量点样器由测微尺控制,也可与Nanomat型点样装置配合,以使点样位置。
电动点样装置*适用于上述各种点样器的配套使用,并且还可以自动控制点样次数,点样器在薄居上的停留时间,以及点样器接触薄层的速度。还可避免手持毛细管点样时的用力不匀,由于靠机械控制接触板面,压力恒定。此外,还有可以用于环形展开及向心展开的薄层点样装置。
(二)带状点样
当样品溶液体积大、浓度稀时,采用自动点样设备进行带状点样。定量分析的点样范围为1-99μl,制备型分离点样范围为5-490μl,作定量标准曲线时,可由同一标准溶液,自动点上不同体积的标准液,故快速简便,利用内装的微处理器编序操作,简单易行。使用时样品溶液吸在微量注射器中,点样器不接触薄层,而是用氮气将注射器针尖的溶液吹落在薄层上,薄层板在针头下定速移动点成0-199mm的窄带。带状点样展开后的斑点不仅分辨率明显高于点状点样,而且精密准确,为定量分析提供*条件。
(三)自动点样
全自动点样装置结合了现代的电子及机械技术,能进行点状或带状点样,采用先进计算机编程控制,灵活多用,点样量为10nl-50μl,可随意设定点样规范,可应用于单向、双向、环形或向心色谱展开,高度自动化的点样使定量分析结果准确。点样参数可与Scanner Ⅱ扫描仪的CATS扫描分析软件连用,令定量分析工作达到高度自动化。
(四)接触点样
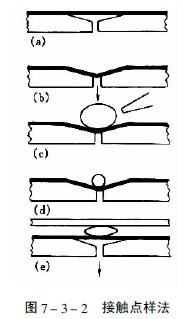
当样品溶液粘度较大或点样体积要超过100μl时,用毛细管点样就有困难,Fenimore设计的接触点样法适用于这类样品的点样,美国制造的Clarke Contact Spotter就是这种接触点样器,见图7-3-2。在带有抽气孔的凹形块的上方覆盖一片涂有疏水性物质的聚氟化物薄膜(a),当减压抽气时薄膜凹陷(b),此时将样品溶液点在凹槽处(c),然后用氮气吹去大部分溶剂(d),再将浓缩后的样品液滴与薄层板的吸附剂层接触(e),此时样品就定量地转移到薄层上。
北京威锋技术开发公司
1988年Kaiser对以上各种点样方式进行了比较,总结其优缺点见表7-3-1、图7-3-3。
表7-3-1 各种点样方式的比较北京威锋技术开发公司
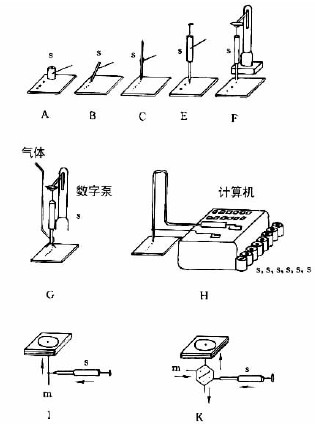
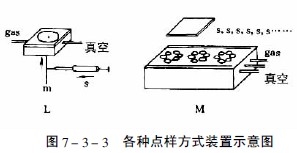
图7-3-3中A-F为zui常用的毛细管或注射器点样:A—一次性数毫米长的薄壁玻管;B—长5-60mm,内径为0.2-0.05mm的熔融石英管,用机械手操作;C—封在玻璃管中的铂铱合金管(100nl,200nl),可点样,生物样品点样后可用火焰清洁,见图7-3-4;E—微量注射器,机械控制,接触板面点样;F—同E,用步进马达控制操作;G—同E,样品溶液用气体喷到薄层板上;H—同E,*用计算机控制点样;I、K、L—借泵系统点样,用于高压平面液体色谱(HPPLC);M—为Fenimore的固态样品环形转移器。
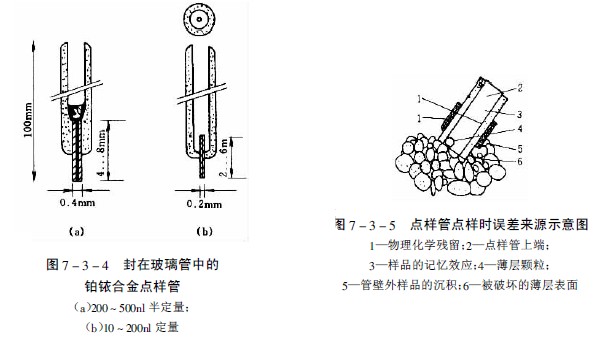
用管状点样器上样的误差来源是由于点样时薄层表面的损伤,溶液“爬壁”效应使外壁样品的沉积、样品的记忆效应以及管中的微小气泡等,见图7-3-5。
点样误差的大小与样品的溶解度,溶液的表面张力,溶剂的粘度、挥发性和极性、点样所耗费的时间、薄层板的质量(粒度、均匀度、薄层厚度、粘合剂的性质)等有关。
(五)特殊的点样技术
在进行植物成分薄层分离之前,提取是首要的步骤,常用适当溶剂进行待测物质的提取,但此法操作麻烦、样品需要量较大,浓缩提取液时对热不稳定的成分易破坏,因此人们研究了样品不经提取而直接点样的方法。
1.热微量抽出法
半个世纪前,人们将一些药物进行微量升华,对升华物进行晶型、熔点以及理化性质等鉴定。在zui简单的情况下,只需将几毫克生药粉末放在微量载玻片上加热,升华物被收集到距离放样品载玻片1mm的另一块载玻片上。1968年Stahl将热微量分离转移技术与薄层色谱法连用,称为“热微量抽出法”此法的优点是大大缩短了提取生药中挥发性成分前处理的时间,简化了提取方法,减少了样品用量,缺点是不宜用于非挥发性成分。
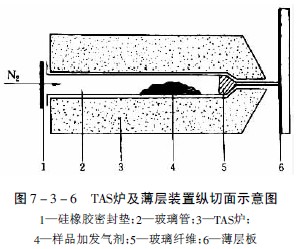
将植物粉末样品放在一端拉细的玻璃管中,并同时放入含有一定水分的合适添加剂,使加热过程中保持连续的水蒸气流(在TAS法中除用含水的分子筛作中性发气剂外,还可用酸性发气剂,如丙二酸,在180℃时产生二氧化碳和乙酸,能使样品中的碱性挥发性成分以盐的形式保留而不逸出,而酸性挥发性成分随酸性气体转移到薄层上;反之亦可用碱性发气剂,如含氨的氯化钙,使样品中的碱性成分随氨气转移到薄层上),管的一端用硅橡胶薄膜严密封闭,并放置在一定温度的TAS炉上,装置见图7-3-6。
加热至220℃,挥发性成分气化,并从玻璃管一端的毛细管口逸出,管中同时以30ml/min的氮气流帮助挥发性成分逸出,逸出的气化成分点加到离出口处1mm的薄层上,待上样完毕,按常规方法展开,即可获得热馏分色谱图。
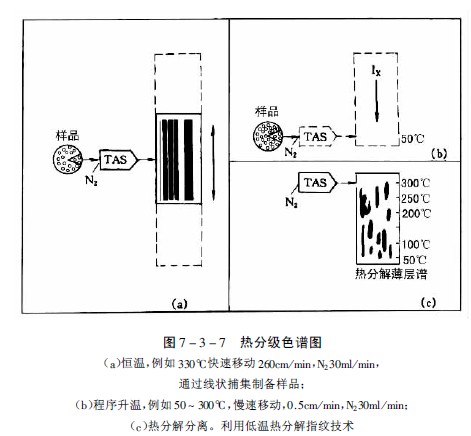
Stahl和Lott发展了此法,设计了可以一定速度横向移动的薄层板装置,可使受热后挥发性成分或热分解产物附着在移动的薄层板起始线上,加热方式可以恒温或程序升温,从而可以得到各种样品的特征谱,改进后的方法称为热分级色谱法。图7-3-7为用市售的“TASOMAT”装置得到的特征图谱。
这种微量快速的上样方法,对天然化合物及合成高分子化合物的分离和鉴定已成为一个重要的手段。下村等在中药厚扑鉴别的研究中应用了此法,只需少量样品即可鉴别厚扑酚(honokiol);桥本庸平等用此法分离了的旋光异构体,管内残渣经生物碱检查呈阴性,证明加热后的挥发是*的。Jolliffe等将含有某些生物碱的药物,如乌头、槟榔、颠茄、金鸡纳、麦角、曼陀罗、天仙子、萝芙木、番木鳖等31种植物药进行热微量抽出法,逸出的挥发性成分直接点在薄层上,用五种不同展开剂展开,用罗丹明B及奶油黄为参比物,显色后对所分离的生碱进行鉴定。Cheml等将金鸡纳树皮中的生碱在炉温300℃时挥发至硅胶板上,展开后鉴定辛可宁等七种生碱。Van Meer等成功地用本法分离了缬草根中的缬草素及异缬草素。Liptak等用本法检验洋茴香、洋甘菊和欧薄荷以及它们的精油类成分。
中成药组成复杂,药典收载的中成药薄层色谱鉴定法均系用溶剂提取后和单味药材或单一成分进行对照。曾美怡等试用热微量转移法鉴定中成药,方法简述如下:
成方制剂的处理:蜜丸拌以等量硅胶5;片剂研成细末;胶囊剂和散剂不加处理;油剂可点在硅胶板上,然后将硅胶连油刮下。
热微量转移条件将样品装入用铝箔卷成的细筒,加一粒含水分子筛,置加入管内,于220-225℃加热60-90s。样品中的挥发性、热分解或升华性成分即直接转移至薄层上。
定位用茴香醛试剂或用荧光法。
样品用量成方制剂为10-16mg,单味药为4-8mg。受试样品有冠心苏合丸、复方丹参片、七厘散、银翘解毒丸、六味地黄丸等成方及单味药,进行了成功的探索,其优点是不需经过复杂的前处理即可得到可供鉴别的色谱。此外,本法也可以应用于小量生药的半定量分析,生药成分的微量制备以及生药的指纹鉴定等。
2.流体提取法
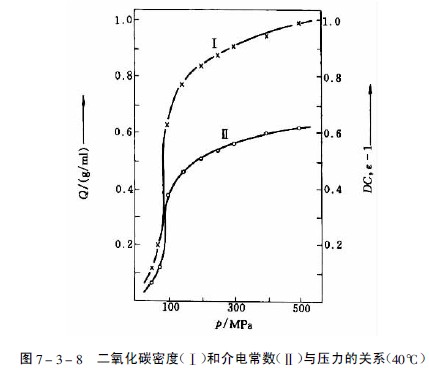
在临界压力和临界温度以上相区内的气体为超临界流体,此时气体的密度、介电常数和溶剂样性质均显着增加,见图7-3-8。物质在超临界流体中的溶解度由于压缩气体与溶质分子间相互作用加强而大大增加。
改变温度和压力就可改变超临界流体的特性,因此可以分别在一定温度和压力条件下将不同极性的成分从样品中提取出来,这种方法与薄层色谱联用,称流体提取一薄层色谱法(Fluid extraction-TLC,FE-TLC)。
用于这种提取过程的气体有二氧化碳、氧化亚氮、乙烯、三氟甲烷、六氟化碳、氮气、氩气等。表7-3-2中列出了各种气体在临界点的物理参数。其中二氧化碳是zui常用的一种气体。
表7-3-2几种气体的物理参数
北京威锋技术开发公司
图7-3-9为FE-TLC示意图,从高压气瓶1出来的气体,通过电动隔膜压缩器6压至7.0-40MPa,压缩器和微量萃取器12在恒温器内保持恒温。萃取器的体积为2ml,被萃取样品置在其中。先关闭阀门11,待高压提取器内气压达到提取压力,提取过程达到平衡后即打开。超临界流体通过一毛细管喷出。由于气流压力下降至大气压,带出的提取物析离并吸附在薄层板13上,薄层板与毛细管出口间距离为1-5mm。点样后按薄层常规方法操作。通过分步提高提取压力,可以分步提出不同成分。
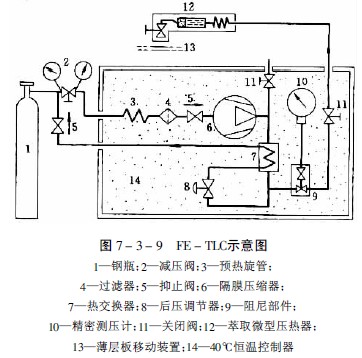
Stahl以二氧化碳为提取气体,研究了不同化合物的可提取性与化学结构的关系。实验结果说明,烃类和亲脂性化合物,如酯、醚、内酯和氧化物等,气压达7.0-10.0MPa即可提取,结构中引入—OH或—COOH基,则提取困难,对于多羟基酚、多酚基酸、糖和氨基酸等在500个atm下也不能提出。
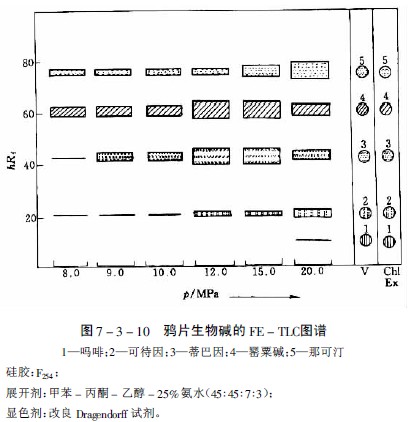
在研究萝芙木、长春花、奎宁和等含27种生物碱的提取性时,选出氧化亚氮为适宜的提取气体,但提取能力随生物碱而异。如分离中主要生物碱时,在约8PMa下和那可汀很易提出,蒂巴因和可待因则较难提出,而吗啡即使压力达20MPa也仅提出极少量。提取一薄层色谱见图7-3-10及图7-3-11。
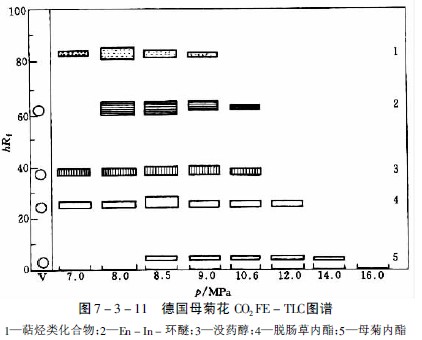
母菊内酯是德国母菊花的有效成分,极易受热分解产生蓝色的菊奥 ,因而无法用水蒸气蒸馏获得。用溶剂提取则得率低,如用二氧化碳超临界流体提取,通过部分提取的方法,可得到母菊内酯及挥发性含氧化合物,经硅胶柱色谱分离即可得到母菊内酯结晶。
北京威锋技术开发公司
